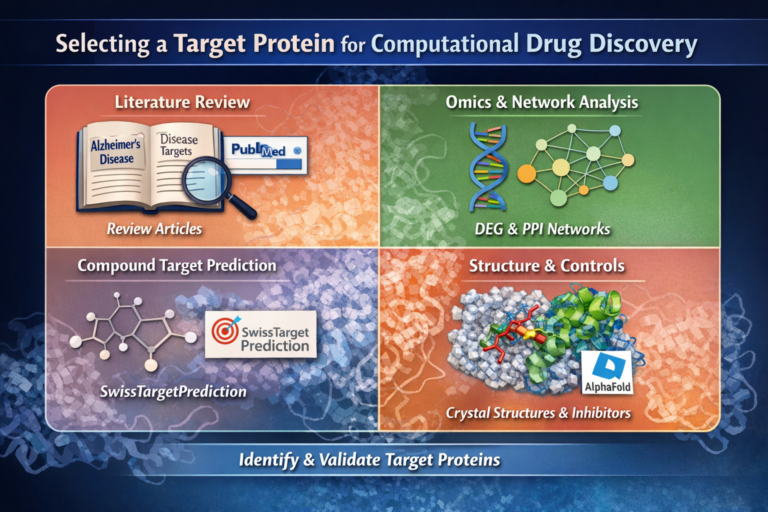
Structure-Based Drug Discovery: A Case Study with RIPK1
Overview
Structure-based drug discovery (SBDD) is a widely used computational approach that leverages the three-dimensional structure of biological targets to identify and optimize potential drug candidates. In this introductory tutorial, we present a real-world case study using Receptor-Interacting Protein Kinase 1 (RIPK1)—a clinically relevant target involved in inflammation, neurodegeneration, and cell death pathways.
This post is designed for students and early-career researchers who are interested in computational drug discovery and want to understand the overall workflow without being overwhelmed by technical complexity. Future posts from Idgence Research will provide step-by-step hands-on tutorials covering each stage in detail.
Why RIPK1?
RIPK1 is a serine/threonine protein kinase that plays a central role in regulating cell survival, inflammation, and necroptosis. Dysregulation of RIPK1 has been linked to several diseases, including neurodegenerative disorders and inflammatory conditions.
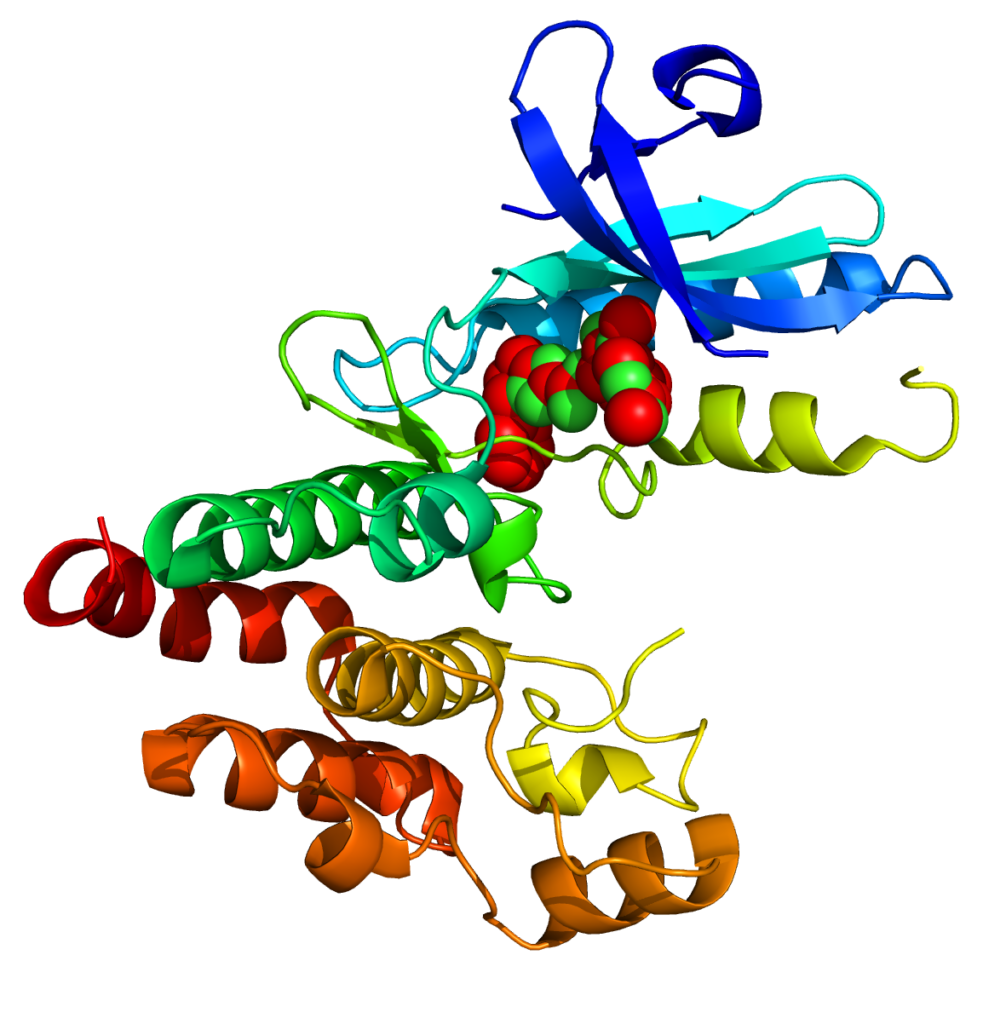
- UniProt entry: Receptor-interacting serine/threonine-protein kinase 1 (UniProt ID: Q13546)
https://www.uniprot.org/uniprotkb/Q13546/entry - AlphaFold structure:
https://alphafold.ebi.ac.uk/entry/Q13546
RIPK1 has also been the focus of multiple peer-reviewed computational studies, making it an excellent example for demonstrating structure-based drug discovery workflows.
Case Study Background
In previous studies, we investigated RIPK1 inhibition using computational approaches such as virtual screening, molecular docking, and molecular dynamics simulations:
- Virtual screening of flavonoids as potential RIPK1 inhibitors for neurodegeneration therapy
Bepari et al., PeerJ (2023)
https://peerj.com/articles/16762/ - A computational study to target necroptosis via RIPK1 inhibition
Bepari et al., Journal of Biomolecular Structure and Dynamics (2022)
https://www.tandfonline.com/doi/abs/10.1080/07391102.2022.2108900
These studies form the scientific foundation for the simplified workflow presented here.
Protein Structure Preparation
A critical requirement for structure-based drug discovery is a reliable three-dimensional structure of the target protein.
- We initially downloaded the X-ray crystal structure of human RIPK1 from the RCSB Protein Data Bank (PDB ID: 6NYH, resolution 2.10 Å).
- This structure contains unmodelled and partially modelled residues, which can affect downstream analysis.
- To address this, a template-based homology model of the RIPK1 kinase domain was generated using the SWISS-MODEL webserver:
https://swissmodel.expasy.org/
Future tutorials will explain protein structure cleaning, validation, and refinement in detail.
Key Steps in Structure-Based Drug Discovery
1. Target Identification
Select a disease-relevant protein with biological and clinical significance (e.g., RIPK1).
2. Protein Structure Preparation
Obtain and refine a reliable 3D structure using experimental data or homology modeling.
3. Ligand Selection
Compile small molecules from natural products or chemical libraries for screening.
4. Molecular Docking
Predict how ligands bind to the protein’s active site using docking software.
- AutoDock Vina 1.2.0 is a widely used docking tool:
https://pubs.acs.org/doi/abs/10.1021/acs.jcim.1c00203
5. Molecular Dynamics Simulation
Evaluate the stability of protein–ligand complexes over time.
- GROMACS is a high-performance molecular simulation package:
https://www.sciencedirect.com/science/article/pii/S2352711015000059
6. Result Interpretation
Analyze binding modes, stability, and interaction patterns to prioritize candidates.
Learning and Research at Idgence Research
Idgence Research integrates computational tools, interdisciplinary expertise, and training to support modern scientific discovery.
- Training programs: https://idgence.org/training
- Research initiatives: https://idgence.org/research
- Our team: https://idgence.org/people
- Contact us: https://idgence.org/contact
What’s Next?
This post provides a conceptual overview of structure-based drug discovery using RIPK1 as an example. Upcoming tutorials from Idgence Research will cover:
- Protein preparation (hands-on)
- Docking workflows with AutoDock Vina
- Molecular dynamics simulations using GROMACS
- Binding free energy analysis
- Best practices and common pitfalls
Stay connected with Idgence Research to explore these topics step by step.